A system for analyzing bacterial RNA-seq data
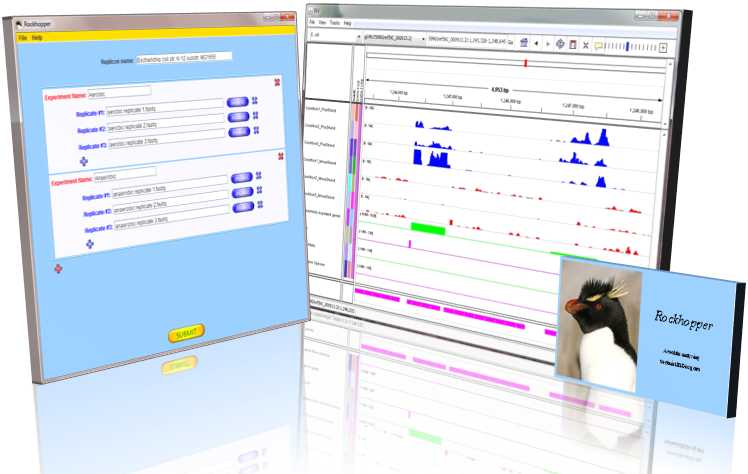
Rockhopper is a comprehensive and user-friendly system for computational
analysis of bacterial RNA-seq data.
As input, Rockhopper takes RNA
sequencing reads output by high-throughput
sequencing technology (FASTQ, QSEQ, FASTA, SAM, or BAM files). Rockhopper supports the following tasks:
- Reference based transcript assembly (when one or more reference genomes are available)
- Aligning reads to genomes
- Assembling transcripts
- Identifying transcript boundaries and novel transcripts such as small RNAs
- De novo transcript assembly (when reference genomes are unavailable)
- Normalizing data from different experiments
- Quantifying transcript abundance
- Testing for differential gene expression
- Characterizing operon structures
- Visualizing results in a genome browser
References
If you make use of Rockhopper in your work, please cite the following:
A computational system for identifying operons based on RNA-seq data. Brian Tjaden.
Methods, 176:62-70, 2020.
De novo assembly of bacterial transcriptomes from RNA-seq data. Brian Tjaden. Genome Biology, 16:1, 2015.
Computational analysis of bacterial RNA-seq data. Ryan McClure, Divya Balasubramanian, Yan Sun, Maksym Bobrovskyy, Paul Sumby, Caroline A. Genco, Carin K. Vanderpool, and Brian Tjaden. Nucleic Acids Research, 41(14):e140, 2013.
 |
This material is based upon work supported in part by the National Science Foundation under grant No. 0919808 and by the National Institutes of Health under grant GM102755. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the author(s) and do not necessarily reflect the views of the National Science Foundation or the National Institutes of Health. |
 |