Reading
How to turn in this Project
You are required to turn in both a hardcopy and a softcopy. If working as a team, the team should submit a single hardcopy and a single softcopy (to either team member's account). Please make sure to keep a copy of your work, on your computer, in your private directory (or, to play it safe, both).Hardcopy Submission
Your hardcopy packet should consist of:- The cover page;
- Your modified
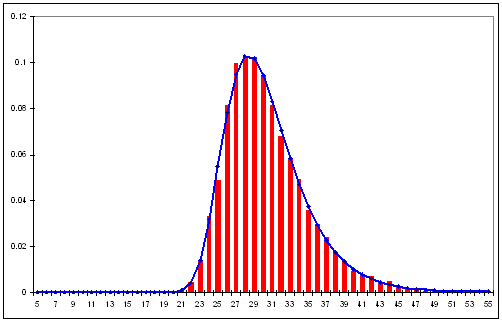
Alignment.javafile from Task 1. - A single graph containing both a histogram of normalized random alignment scores and a plot of the corresponding extreme value distribution from Task 1.
Softcopy Submission
You should submit your final version of your
Alignment directory to the
drop/project3 directory in your
account on the CS server.
Your friend's biology professor has sent you an email asking to see you in her office. Uh oh, you think. This can't be good. You didn't do anyone's homework for them, you just developed some software to automate a few redundant tasks for your fellow students. If anything, the professor should be thanking you for providing a tool that minimizes students' "busy" work thereby leaving more time for creative and critical thinking. You arrive at the professor's office ready to defend yourself from the wild and unfair accusations when the professor says that she is impressed by your computational prowess and wants to know if you would be interested in joining her research group. Members of her lab are constantly comparing genomic sequences to each other searching for similarities and relationships and the professor thinks that the lab's investigations could be improved by someone with computational skills. Delighted by the opportunity, you tell her that you'll go straight to the Registrar's office to sign up for an independent study with her. She tells you that the opportunity is not for course credit. Oh, well you haven't maxed out the hours for your job on campus, so you can pick up a second job working in her lab. She tells you that the opportunity is not for hourly pay. Oh, well the intellectual benefits from engaging in research are much more valuable than any academic credit or financial remuneration. The professor nods and smiles astutely.
Task 1: Optimal Pairwise Alignment
We are providing you with a starter program that computes the optimal
global alignment of two genomic sequences. In this problem, you will
extend the starter program so that it computes more sophisticated
pairwise alignment operations. The starter program,
Alignment.java is stored in the
/home/cs313/download/Alignment subdirectory on
the CS server. You can immediately compile and execute the existing
program. The main method contained in the Alignment.java
file assumes that the user has entered the names of two FASTA files in the command
line when executing the program, for example:
java Alignment data/dna_seq1.txt data/dna_seq2.txt
The Alignment program will compute the optimal global pairwise alignment
of the two genomic sequences found in the two FASTA files. For the sequences in the
two example files mentioned above, dna_seq1.txt and dna_seq2.txt,
the Alignment program should report an optimal global alignment score of 6 and
an optimal global alignment of:
CGGCTTG
|| ||
AGG-TTC
The program also reports a p-value of 9999.9, which is not the corrent p-value. One of your tasks, described below, will be to modify the program so that it reports a correct p-value.
To experiment with a sample solution, we have provided you with a file Alignment_Sols.class. To execute the sample solutions, for example, using a local alignment, with matrix scoring, and affine gaps, you could include the following code in the main method of your Alignment.java file:
Alignment_Sols a_sols = new Alignment_Sols(new File(args[0]), new File(args[1]));
a_sols.setLocalAlignment();
a_sols.setMatrixScoring("data/DNA_Matrix.txt");
a_sols.setAffineGaps(-11, -2);
a_sols.computeAlignment();
a_sols.calculatePValue();
System.out.println(a_sols);
The above code, which invokes the sample solutions, might cause the following to be output:
|
You should study the starter code in the Alignment.java file.
The starter code implements most but not all of
the methods described in the Alignment contract found here.
You will be implementing a number of additional methods as described below.
- While the starter code supports computation of an optimal global
alignment of two sequences, you must write code that supports computation of
an optimal local alignment of two sequences. In order to compute
the optimal local alignment of two sequences, you must fill in the
following two method skeletons:
private void localAlignment()
Computes the optimal local pairwise alignment score for two genomic sequences. An alignment table (i.e., 2D integer array) is populated as part of the computation of the optimal alignment score. A backtracking table (i.e., 2DPointarray) is also populated so that the optimal alignment (and not just the score) can be re-constructed using the methodlocalBacktrack.private String localBacktrack()
Once an alignment score has been computed, the backtracking table is searched to determine the optimal alignment. The optimal local alignment is re-contructed by searching through the backtracking table. The optimal alignment is returned as aString.When coding for optimal local alignments, you may find the methods
alignmentTableToStringandbacktrackTableToStringuseful for debugging your code. You are free to use any additional methods and variables that you deem appropriate. - The starter code supports only fixed match/mismatch scoring. In
fixed scoring, there are no gradations of similar or dissimilar
pairs of characters. Rather, all aligned pairs of identical characters are
given one score (e.g., 5) and all aligned pairs of non-identical characters are
given one score (e.g., -4). As an alternative to fixed scoring, each pair
of aligned characters can be given a score that may be unique. For example,
an "A" aligned with a "G" may yield a differnt score than an "A" aligned
with a "C". In this alternative scoring model, the scores for all pairs
of aligned characters are often represented as a matrix. For instance,
here is a scoring matrix for DNA sequences
and here is a scoring matrix for protein sequences.
Notice that the two above matrices are symmetric, i.e., aligning character
xwith characteryis equivalent to aligning characterywith characterx. Such a scoring model is known as matrix scoring.You must implement matrix scoring. You will need to read in a matrix file (such as
DNA_Matrix.txtorBlosum62_Matrix.txtfrom thedatadirectory and store the matrix values in an appropriate data structure (the data structure need not resemble a matrix - think about what data structures might be most efficient). Given two characters to be aligned, you will need to support quickly determining the alignment score of the two characters. In order to support matrix scoring, you must fill in the following two methods:public void setMatrixScoring(String fileName)
When aligning two characters, one from each genomic sequence, the alignment score of the two characters should be determined from a matrix of scores found in a file with the specified name. In fixed scoring, all pairs of identical characters (e.g., G|G, C|C, T|T) are scored the same and all pairs of different characters (e.g., G|C, G|T, C|T) are scored the same. However, with genomic sequences, not all pairs of characters are equally similar or dissimilar. In matrix scoring, different pairs of identical characters (e.g., G|G, C|C, T|T) may be scored differently and different pairs of mismatching characters (e.g., G|C, G|T, C|T) may be scored differently. For example, adenine (A) and guanine (G) are both purines whereas cytosine (C) and thymine (T) are both pyrimidines. Since adenine is more similar to guanine than to thymine, an adenine aligned with a guanine (A|G) should not penalize an alignment as much as an adenine aligned with a thymine (A|T). Analogously for protein sequences, two different hydrophobic amino acids aligned together might not penalize an alignment as much as a hydrophobic amino acid aligned with a hydrophilic amino acid. In matrix scoring, the alignment score of every possible pair of characters is specified in a matrix that must be read in from a file. For DNA sequences, since there are 4 characters in the DNA alphabet, there are 16 possible pairs of characters and the matrix contains 16 entries. For protein sequences, since there are 20 characters in the protein alphabet, there are 400 possible pairs of characters and the matrix contains 400 entries.private int getScore(char c1, char c2)
Returns the alignment score for two characters. - The starter code supports only linear gap scoring. In
linear gap scoring, each gap in a run of consecutive gaps is scored
the same amount (e.g., -6). As an alternative, to linear gap scoring,
affine gap scoring scores the first gap in a run of consecutive
gaps with one score, alpha, and subsequent gaps in a run of
consecutive gaps with a second score, beta. Affine gap scoring is meant
to model empirical biological evidence that the existence of a gap is more
significant than the length of the gap. It is expensive, biologically, to
add to or splice from a genomic sequence, but the length of the addition or deletion
is relatively less important than its existence.
You must implement affine gap scoring. In order to support affine gap scoring, you must fill in the following method:
private int getGapScore(int i, int j, boolean isGapAbove)
Returns the gap score for a particular position in a pairwise alignment. The parametersiandjrepresent the indices for sequence #1 and sequence #2, respectively. I.e.,iandjrepresent the row and column in the alignment and backtrack tables that are populated during computation of the optimal alignment. TheisGapAboveparameter indicates, for affine gap scoring, an extension of a gap from a table entry above (as opposed to a gap from a table entry to the left). - The starter code fills in the entire dynamic programming table, considering
any possible combination of gaps, when performing a pairwise alignment. This
requires O(n2) steps when aligning sequences
of length n. If we restrict the number of gaps that we consider
when performing a pairwise alignment, we need only fill in part of the
dynamic programming table. If we restrict the number of gaps to k
then we can fill in the table in O(k * n) steps,
which is linear for small constant values of k.
You must implement fast, linear time pairwise alignment. You need only implement fast, linear time alignment for global alignment, not local alignment. In order to support fast alignment, you must fill in the following method:
private void globalAlignment_FAST()
Computes the optimal global alignment score for two genomic sequences when considering at least a small fixed number of gaps. An alignment table (i.e., 2D integer array) is partially populated as part of the computation of the optimal alignment score. For k gaps, runs in O(k * n) time. A backtracking table (i.e., 2DPointarray) is also partially populated so that the optimal alignment (and not just the score) can be re-constructed using the methodglobalBacktrack. - The starter code does not estimate p-values for an alignment.
The p-value of a given alignment is the probability (between 0.0 and 1.0) that the
optimal pairwise alignment score of two random sequences is greater than or equal to the
optimal pairwise alignment score for the given alignment. A p-value close to 1.0 suggests
that an alignment was likely to have occurred merely by chance. A p-value close to 0.0 suggests
that an alignment was unlikely to have occurred by chance. In this case (especially if the p-value
is less than about 0.05), the alignment is significant and the sequences may be deemed
similar.
You must implement code so that a p-value is estimated for each alignment. In order to compute p-values, you must fill in the following three methods:
private static double getVectorMean(Vector<Integer> v)
Returns the mean (i.e., the average value) of aVectorofIntegers.private static double getVectorStandardDeviation(Vector<Integer> v)
Returns the sample standard deviation of aVectorofIntegers.public void calculatePValue()
Estimates the p-value of an alignment. A p-value for an alignment of two sequences can be estimated as follows. Randomly generate 1000 pairs of sequences by randomly permuting the original two sequences. For each of the 1000 pairs of random sequences, determine the optimal pairwise alignment score. These 1000 scores should be stored in aVectorofIntegerobjects. These 1000 scores approximate an extreme value distribution. Calculate the mean and standard_deviation of the 1000 scores. The mean and standard_deviation can be used to calculate two parameters, mu and beta, representing the extreme value distribution. mu can be calculated as mean - (0.5772*beta). beta can be calculated as standard_deviation * √6 / π. Finally, the p-value can be calculated as 1.0 - e^(-e^(-(x-mu)/beta)) where x is the optimal pairwise alignment score of the original pair of sequences.As a final task, calculate the p-value for two longer sequences (at least 100 characters in length and preferably protein sequences). Your
calculatePValuemethod should have stored in aVectorthe 1000 alignment scores from randomly generated pairs of sequences. Using thisVectorof 1000 alignment scores from random sequences, invoke the methodoutputHistogramOfRandomAlignmentScores. TheoutputHistogramOfRandomAlignmentScoresmethod should output a histogram to a file. The first column in the output file corresponds to 101 different alignment scores. The second column indicates how many of the 1000 alignments resulted in the corresponding score. The third column is a normalized version of the second column, i.e., the number in the second column is divided by the total number of alignment scores. The fourth column represents a mathematical function - an extreme value distribution - that approximates the third column. The extreme value distribution is determined from the mean and standard deviation of the set of 1000 alignment scores from random sequences. The extreme value distribution is used to estimate the p-value for an alignment. As a final step, using your favorite graphing application, create a single graph from the third and fourth columns in this file. The third column should be graphed as a bar graph and the fourth column should be graphed on top as a line graph.
{kind=link}