How to turn in this Project
You are required to turn in both a hardcopy and a softcopy. If working as a team, the teamshould submit a single hardcopy and a single softcopy (to either team member's account). Please make sure to keep a copy of your work, on your computer, in your private directory (or, to play it safe, both).Hardcopy Submission
Your hardcopy packet should consist of:- The cover page.
- Your
HMM.javafile from Task 1. - Your filled in Table Of Results from Tasks 2, 3, and 4.
- Your
Ecoli_states.txtandEcoli_transitions.txtfile from Task 3. - Your
Yeast_states.txtandYeast_transitions.txtfile from Task 3. - Your
Arabidopsis_states.txtandArabidopsis_transitions.txtfile from Task 3. - Your
Fly_states.txtandFly_transitions.txtfile from Task 3. - Your
Ecoli_states.txtandEcoli_transitions.txtfile from Task 4. - Your
Yeast_states.txtandYeast_transitions.txtfile from Task 4. - Your
Arabidopsis_states.txtandArabidopsis_transitions.txtfile from Task 4. - Your
Fly_states.txtandFly_transitions.txtfile from Task 4.
Softcopy Submission
You should submit your final version of your
GeneFinder directory to the
drop/project8 directory in your
account on the CS server.
The phone rings for nearly two minutes before the sound of it brings you to consciousness. You look at the clock... it's 3:00am. Who could possibly be calling at this hour? Hello? Someone from the Mobile Foundation? No, that's the Nobel Foundation. Huh? Oh. Do they know it is 3:00am? They do. It is 8:00am in Stockholm. Oh. So, like, what's up, why are you calling? The Nobel Prize in Physiology or Medicine. Are you serious? They are very serious. Your mind is suddenly lucid. In your email, you are told, is an official letter from the Nobel Foundation congratulating you on your award. There will be a press release in a couple of hours. They are citing your work integrating computational and experimental sciences, your breakthrough research on how cells regulate oncogenes, the computational advances that you championed that have become the gold-standard for identifying genes in a genome, and your lifetime of contributions to science and medicine. Wow, you think. Overwhelmed, your mind races back to your days in college... your CS313 instructor wasn't right about much as you recall, but he did foretell this moment. Weird.
Background
In this project, your goal is to develop a gene-finder based on a hidden Markov model (HMM). In the first task, you will complete an implementation of a HMM. In the second, third, and fourth tasks, you will use the HMM as a gene-finder. Given a genomic sequence, your gene-finder should predict where any protein-coding genes can be found in the genomic sequence. More specifically, your gene-finder should indicate for each nucleotide in the genomic sequence whether the nucleotide is part of a protein-coding region (i.e., a gene) or not.
Once you have developed your gene-finder, you will predict the locations of genes in genomic sequences from several different species. You will then compare your predictions with the locations of known/documented genes in the species to evaluate how accurate your predictions are. The accuracy of your predictions will vary depending on the details of your HMM and on the species for which you are predicting genes (it is easier to identify genes in the genomes of some species than in others).
Task 1: Implementing a Hidden Markov Model (HMM)
We have provided you with a partial implementation of a HMM. The HMM implementation consists of two classes:
- An instance of the
Stateclass represents a single state in a HMM. - An instance of the
HMMclass represents a hidden Markov model.
The contracts for these two classes can be found here.
An implementation for these two classes is stored in the
download/GeneFinder subdirectory on the CS server.
Study the two provided classes, State and HMM.
The HMM class contains code to read in from files information about a HMM:
- The first file read in by the HMM should contain information about
the states of the HMM and their emission probabilities. Each line of the
file contains the name of a state followed by a tab followed by a
comma-delimited list of strings emitted by the state followed by a
tab followed by a comma-delimited list of probabilities of emitting
each string. For examples,
- here is a sample file for a HMM with 3 states
corresponding to 3 coins that each emit
HorTwith some probability. Based on the sample file, the probability of the first coin emittingTis 10% and the probability of the second coin emittingHis 50%. - here is a sample file for a HMM with 3 states
each emitting one or more English letters with some probability. Based on the sample
file, the probability of the first state emitting
Eis 30%, the probability of the second state emittingERis 60%, and the probability of the third state emittingREVERis 100%.
- here is a sample file for a HMM with 3 states
corresponding to 3 coins that each emit
- The second file read in by the HMM should contain information about
the probabilities of transitioning between states of the HMM. The file
contains a tab-delimited matrix (2D array) of probabilities, where
the first row and column correspond to the names of the states. For examples,
- here is a sample file for a HMM with 3 states indicating the probability of transitioning from each state to any other state. Based on the sample file, the probability of transitioning from the first state to the first state is 90%, the probability of transitioning from the second state to the first state is 2%, and the probability of transitioning from the third state to the second state is 30%.
- here is a sample file for a HMM with 3 states indicating the probability of transitioning from each state to any other state. Based on the sample file, the probability of transitioning from the first state to the second state is 45%, the probability of transitioning from the second state to the first state is 85%, and the probability of transitioning from the third state to the third state is 98%.
- The third file read in by the HMM should contain an observation sequence
in FASTA format. For examples,
- here is a sample file
containing a sequence generated by a HMM. The observation sequence
in this sample file was generated by a HMM whose states correspond
to coins that emit
HandT. - here is a sample file containing a sequence generated by a HMM. The observation sequence in this sample file was generated by a HMM whose states emit one or more English letters.
- here is a sample file
containing a sequence generated by a HMM. The observation sequence
in this sample file was generated by a HMM whose states correspond
to coins that emit
The State class does not require any modification.
However, the HMM class is incomplete. The HMM
class does not contain code to determine the optimal annotation
(i.e., parse or state sequence) for an observation sequence. In this task,
you goal is to complete the implementation of the HMM class. To
complete the HMM class, you will need to implement three methods
described in the HMM contract, namely
fillInTableEntry, viterbi, and
determineOptimalAnnotation. These three methods, when completed,
will enable computation of the optimal annotation of an observation
sequence.
With the code available to download on the CS server, we have provided
you with several files in the data_Examples directory
for testing your HMM implementation. We have also
provided you with a sample solution, TEST_HMM,
which you can execute and compare to your own solution. For example,
executing the sample solution with the three provided files corresponding
to a HMM based on 3 coins might yield the following:
|
In the above example, the optimal annotation of the observation sequence HTTHTTTHHT
has a score of -12.192816965761187 and can be achieved by flipping coin 1 once and
then flipping coin 2 nine times.
As another example, executing the sample solution with the three provided files corresponding to a HMM based on three states emitting English letters might yield the following:
|
In the above example, the optimal annotation of the observation sequence
REMEMBEREVERREVERSEMEREVERBS has a score of -38.6211867437036 and can be
achieved by
starting in state 1 and emitting the character R,
transitioning to state 1 and emitting the character E,
transitioning to state 2 and emitting the character ME,
transitioning to state 1 and emitting the character M,
transitioning to state 1 and emitting the character B,
transitioning to state 1 and emitting the character E,
transitioning to state 3 and emitting the character REVER,
transitioning to state 3 and emitting the character REVER,
transitioning to state 1 and emitting the character S,
transitioning to state 1 and emitting the character E,
transitioning to state 1 and emitting the character M,
transitioning to state 2 and emitting the character ER,
transitioning to state 1 and emitting the character E,
transitioning to state 1 and emitting the character V,
transitioning to state 2 and emitting the character ER,
transitioning to state 1 and emitting the character B,
transitioning to state 1 and emitting the character S.
Task 2: Gene Finding (on one strand of a genome)
In this task, you will use the HMM from Task 1 to
predict genes in genomic sequences. Even if you didn't complete Task 1,
you can still complete Task 2 by using the provided sample solution HMM
implementation, HMM_Sols.
Consider the four state HMM shown in the image below:
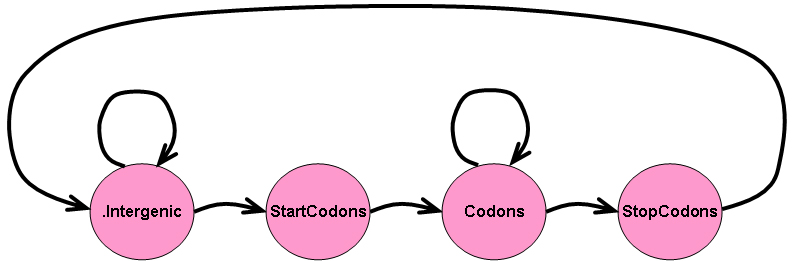
In the above HMM, the .Intergenic state corresponds
to intergenic (non-coding) regions of a genome, the
StartCodons state corresponds to the start codons of
genes (usually ATG but occasionally others such as
TTG,
CTG, GTG, ATT, CGA,
ACG, or AGT, depending on the organism),
the Codons state corresponds to the 64 codons found in genes
(not counting the start or stop codons of the genes), and the
StopCodons state corresponds to the stop codons of genes
(usually TAA, TGA, and TAG, but
occasionally others such as AGA, AGG, or
GGA, depending on the organism).
Suppose we want to use the above HMM to predict genes in a genomic
sequence from the bacteria Escherichia coli. First, we would
need to determine the parameters of the HMM. For example, what is the
probability of emitting different nucleotides from each of the four
states? If we have reason to believe that the GC-content of intergenic
regions in E. coli is about 44%, we might set the probability
of emitting nucleotide G or C in the
.Intergenic state to be 0.22 each, and the probability of emitting
nucleotide A or T to be 0.28 each. Similarly,
if we have reason to believe that the stop codon used by E. coli
is TAA for 65% of its genes, TGA for 28% of its
genes, and TAG for 7% of its genes, then we might set the
probability of emitting nucleotides TAA, TGA,
and TAG in the StopCodons state to be
0.65, 0.28, and 0.07, respectively. The file
Ecoli_states.txt
specifies one possible set of reasonable emission probabilities
for each of the four states. We would also need to determine the
probability of transitioning from each of the four states to
any other of the four states. For example, since approximately 99.4% of
intergenic nucleotides in the E. coli genome are followed
by another intergenic nucleotide and only 0.6% of intergenic
nucleotides are followed in the genome by a protein-coding nucleotide
(i.e., the first nucleotide of a gene's start codon), we might set
the probability of transitioning from the .Integenic
state to the .Intergenic state to be 0.994 and
the probability of transitioning from the .Intergenic
state to the StartCodons state to be 0.006. The file
Ecoli_transitions.txt
specifies one possible set of reasonable transition probabilities
for the four states.
Once parameters of the HMM are determined, for a given genomic
sequence (i.e., observation sequence), the HMM can then be used
to predict genes in the genomic sequence by computing
the optimal annotation of the genomic sequence. Any nucleotides emitted
from one of the three codon states in the optimal annotation are predicted
to be protein-coding gene regions and any nucleotides emitted from the
.Intergenic state in the optimal annotation are predicted
not to be protein-coding gene regions. For instance, if we want
to predict whether there are any genes in the first 290 nucleotides
of the E. coli genome, as specified in the file
Ecoli_observation_290.txt,
we can use the HMM to determine the optimal annotation of this 290
nucleotide observation sequence. Based on the model parameters
specified in the files
Ecoli_states.txt and
Ecoli_transitions.txt,
the optimal annotation of the 290 nucleotide observation sequence is:
|
In the optimal annotation above, nucleotides with a period, ., below them
correspond to the .Intergenic state (note that the first character in the
name of the .Intergenic state is a period). Nucleotides with the letter S
below them correspond either to the StartCodons or StopCodons
state (the names of both states start with the letter S), and nucleotides
with a C below them correspond to the Codons state. In the case
of the optimal annotation above, the predicted protein-coding nucleotides correspond
precisely to those of a documented gene, the thr operon leader peptide,
in the E. coli genome known to reside between nucleotides 190 and 255 in the
genome.
Of course, we could predict genes (i.e., determine the optimal annotation) in much longer genomic sequences (i.e., observation sequences) as well. We could predict genes throughout the E. coli genome. If we want to predict genes in genomic sequences from other organism besides E. coli, we can do so as long as we have reasonable sets of model parameters (emission probabilities and transition probabilities). The model parameters should be genome specific because different genomes have different GC-contents, have different affinities for codons, and have different densities of genes within their genomes.
With the code available to download on the CS server, we have provided
you with several files in the data_OneStrand directory. These
files contain reasonable model parameters (emission and transition
probabilities) for E.coli, for Yeast, for
Arabidopsis, and for Fly genomes. Using these files
and the observation sequence files in the data_OneStrand
directory, you should predict genes throughout:
- the genome of E. coli
- chromosome 4 of the Yeast genome
- the first 900,000 nucleotides of chromosome 4 of the Arabidopsis genome
- chromosome 4 of the Fly genome
By default, the HMM application outputs the optimal annotation
of an observation sequence to the screen as well as to a file
named annotation.txt. For large genome sequences, your program may
be slow to output the annotation to the screen, so you may want to comment out the lines
in the main method that print the annotation to the screen (you only
really need the file, annotation.txt).
Once you have predicted genes in a genomic sequence, it would useful
to evaluate how accurate the predictions are. In the data_OneStrand
directory that you downloaded, there are files listing the known genes in
E. coli, Yeast, Arabidopsis, and Fly.
The provided application EvaluateGenePredictions will allow
you to compare your gene predictions to the known genes and assess the
accuracy of your predictions. You can find the contract for the
EvaluateGenePredictions
class here. The EvaluateGenePredictions class reports
the sensitivity and specificity of predictions. The sensitivity is
the percent of documented coding nucleotides that are correctly predicted
as coding nucleotides. The specificity is the percent of documented
non-coding (i.e., intergenic) nucleotides that are correctly predicted as
non-coding nucleotides.
In the first four rows of the Table of Results, please fill in the sensitivity and specificity of your gene predictions based on the four-state HMM for E. coli, Yeast, Arabidopsis, and Fly.
Task 3: Gene Finding (on both strands of a genome)
The four-state HMM described in Task 2 only predicts genes on one strand of a genomic sequence. Of course, to predict genes on both strands of a genomic sequence, we could always predict genes on one strand of the sequence and then create a second sequence that is the reverse complement of the first and then predict genes on the second sequence. However, a better approach may be to use a HMM that predicts genes along both strands simultaneously.
Consider the seven state HMM shown in the image below:
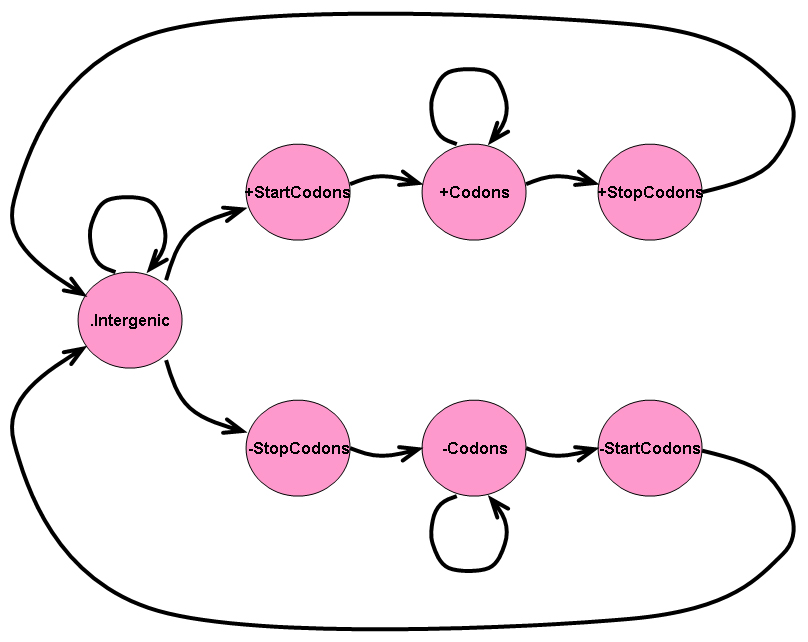
If we were searching for genes along one strand, call it the "+" strand, of a genomic sequence, we would look for a start codon followed by a series of codons followed by a stop codon. If we were searching for genes that resided on the opposite strand, call it the "-" strand, of a genomic sequence, we would look for the reverse complement of a stop codon followed by the reverse complement of a series of codons followed by the reverse complement of a start codon. The seven state HMM above is meant to reflect a HMM that can search for genes along both strands of a genomic sequence simultaneously.
Using the model parameter (emission and transition probabilities)
files in the data_OneStrand directory as a starting
point, you should create new model parameter (emission and
transition probabilities) files corresponding to the seven
state HMM depicted above. You should create new model parameter
files for E. coli, for Yeast, for
Arabidopsis, and for Fly. You should store these
new files in the directory data_TwoStrand. Once you
have created these model parameter files, you should predict
genes throughout:
- the genome of E. coli
- chromosome 4 of the Yeast genome
- the first 900,000 nucleotides of chromosome 4 of the Arabidopsis genome
- chromosome 4 of the Fly genome
Use the EvaluateGenePredictions application to assess
your gene predictions. In rows five through eight of the
Table of Results, please fill in the
sensitivity and specificity of your gene predictions based on the
seven-state HMM for E. coli, Yeast,
Arabidopsis, and Fly.
Task 4: Gene Finding (on both strands of a genome with introns)
The seven-state HMM described in Task 3 only predicts genes that have continuous coding regions. Such a model will work well for bacteria, which do not have introns and exons as part of their genes. However, for eukaryotes, genes are often split into protein-coding exons and non-coding introns. Ideally, we would like our HMM to be able to predict genes that contain exons and introns.
Consider the thirteen state HMM shown in the image below:
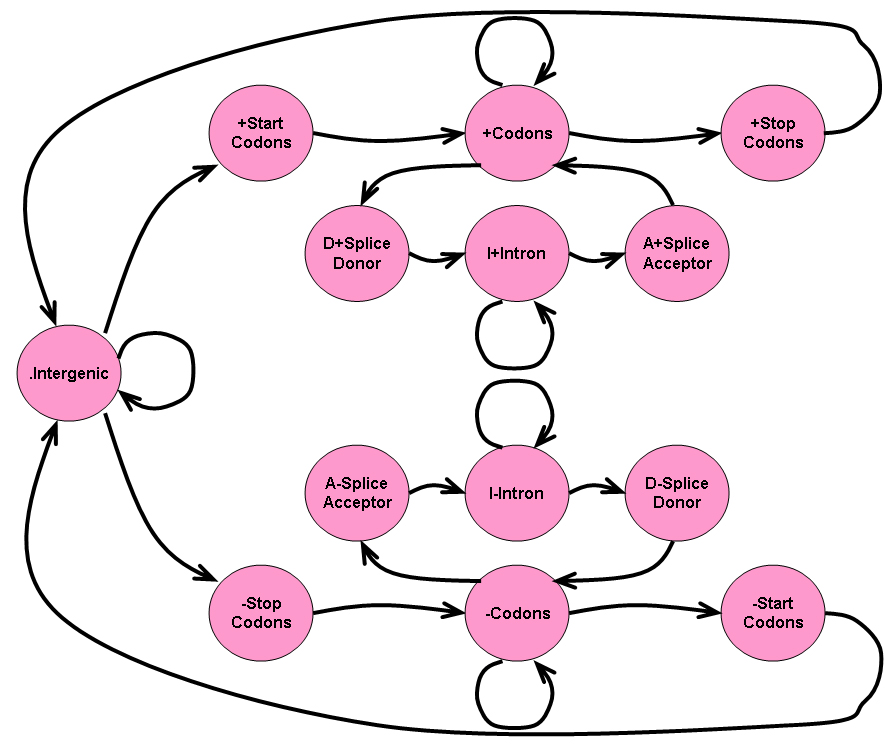
The HMM depicted above is meant to model genes that may have exons and introns. In the model above, a coding region (a series of codons) may be broken up by introns (sequences of non-coding nucleotides).
For the model above, we will make a few simplifying assumptions:
- All introns begin with the splice donor site
GT. - All introns end with the splice acceptor site
AG. - Since introns contain non-coding nucleotides, we will assume the distribution of nucleotides in introns is the same as the distribution of nucleotides in intergenic regions.
- In E. coli, the average length of introns is 0 nucleotides and the average number of introns per gene is 0.0.
- In Yeast, the average length of introns is 150 nucleotides and the average number of introns per gene is 0.04.
- In Arabidopsis, the average length of introns is 150 nucleotides and the average number of introns per gene is 3.8.
- In Fly, the average length of introns is 800 nucleotides and the average number of introns per gene is 2.5.
For this task, your goal is to create new model parameter (emission and
transition probabilities) files corresponding to the thirteen
state HMM depicted above. You should create new model parameter
files for E. coli, for Yeast, for
Arabidopsis, and for Fly. You should store these
new files in the directory data_TwoStrandIntron.
When developing your model parameter files, think about how
you can use the simplifying assumptions described above. As an example
to help you think about the transition probabilities out of a state:
- Consider the three transitions out of the +Codons state. Let us suppose, for example, that genes in a particular genome contain 500 codons on average, not counting the start and stop codons. Let us also suppose that genes in the genome average 5 introns per gene. Then 1 out of the 500 codons in a gene (the last one) would be followed by a Stop Codon, 5 out of the 500 codons would be followed by the start of an intron (D+Splice Donor), and 494 out of the 500 codons would be collowed by another +Codon. Thus, if we know the average number of codons per gene, we can use the above logic to calculate the transition probabilities out of the +Codons state. Do we know the average number of codons per gene in each genome? If not, can we calculate it if we know the transition probabilities for the Codons state?
Once you have created the model parameter files, you should predict genes throughout:
- the genome of E. coli (when running your program on the genome of E. coli it is possible that your program will require more heap memory than is allocated by Java by default, in which case you can increase the amount of heap memory available to your program, e.g., instead of executing "java HMM ..." you can execute "java -Xmx1200m HMM ...")
- chromosome 4 of the Yeast genome
- the first 900,000 nucleotides of chromosome 4 of the Arabidopsis genome
- chromosome 4 of the Fly genome
Use the EvaluateGenePredictions application to assess
your gene predictions. In rows nine through twelve of the
Table of Results, please fill in the
sensitivity and specificity of your gene predictions based on the
thirteen-state HMM for E. coli, Yeast,
Arabidopsis, and Fly.